«оптимисты». как живут люди с орфанными заболеваниями в россии?
Содержание:
- Что такое орфанные заболевания
- Самая частная из редких болезней
- ТОЧНАЯ И ОПЕРАТИВНАЯ ДИАГНОСТИКА – ПУТЬ К СПАСЕНИЮ ЖИЗНИ
- Хадассе нет равных в диагностике орфанных заболеваний
- Орфанные заболевания: перечень
- БЛАГОТВОРИТЕЛЬНЫЕ ОРГАНИЗАЦИИ
- Цена здоровья
- Редкие генетические заболевания
- Лечение без СМС
- Ваня выздоровел
- Методы диагностики адреногенитального синдрома
- Диагностика орфанных заболеваний
- Один на десять тысяч
- Немного о деньгах
- Персональные клинические испытания
- Один из многих тысяч
- Причины адреногенитального синдрома
- Лечение редких заболеваний (методы)
- Орфанные заболевания: статистика
- Вопрос денег
- Примеры орфанных заболеваний, которые диагностируют и лечат в МЦ Хадасса
Что такое орфанные заболевания
Термины «орфанные заболевания» или «болезни-сироты» (от англ. orphan — сирота) появились в 1983 году, когда в Америке был принят закон о препаратах-сиротах («Orphan Drug Act»), благодаря которому поощрялись фармацевтические компании за разработку лекарственных средств с целью лечения редких патологий
В России на эту проблему обратили внимание только в 2011 году и закрепили права пациентов с редкими заболеваниями в законе «Об охране здоровья граждан»
Сегодня понятие орфанных (редких) болезней закреплено в медицинской терминологии законодательством многих стран. В группу орфанных болезней входят малоизвестные генетические, аутоиммунные и инфекционные патологии, а также редкие формы рака, аномалии и синдромы. Орфанные заболевания, вызванные мутациями генов, характеризуются тяжелым хроническим течением и требуют длительного дорогостоящего лечения. Другие виды редких патологий также ухудшают качество жизни, быстро прогрессируют, могут приводить к инвалидности и смертельному исходу. Большинство орфанных болезней у детей и взрослых являются малоисследованными состояниями, для которых еще не разработаны методы терапии. Кроме того, не часто удается диагностировать подобные заболевания своевременно.
Самая частная из редких болезней
Сам того не понимая, с утра до ночи малыш борется за жизнь. Корсет и туторы поддерживают мышцы. Эти неудобные «доспехи» нужно носить постоянно. Дышать свободно удаётся лишь благодаря аппарату неинвазивной вентиляции легких.

На мучения мальчика обрекла спинальная мышечная атрофия 1 типа – это самая частая из редких болезней.
«Сейчас ребенку 8 месяцев, он не поднимает ноги, не переворачивается, не держит головку. Врачи говорят, что начала деформироваться грудная клетка. Такие детки не доживают до двух лет», – рассказывает мама Матвея Анна Безуглая.
«Перед тем, как планировать семью, надо сдать анализы на СМА. Если подтверждается мутация у матери и отца, то на 9-12 неделе проводятся исследования и берутся анализы у ребенка. При выявлении болезни надо сразу ввести препарат», – разъясняет папа Матвея Александр Куликовский.

Против СМА есть всего несколько лекарств. Одно из них – «Спинразу» – мальчик уже принимает: его бесплатно предоставляет государство, но оно, считают родители, только тормозит болезнь. Надежда Куликовских – на новую разработку.
Производитель обещает – достаточно одной инъекции. Только цена её баснословная – 2 миллиона долларов. Препарат признан книгой рекордов Гиннесса самым дорогим в мире. К тому же, в России он не зарегистрирован. Купить лекарство регион имеет право – при определенных обстоятельствах.
«Если будет показано, если все факторы совпадут: хотят родственники, хотят врачи, все оценили риски, мы будем добиваться того, чтобы государство выделило эти деньги. Должен быть федеральный консилиум, обращение соответственно», – говорит первый заместитель председателя Комитета по здравоохранению Санкт-Петербурга Андрей Сарана.

Куликовские ждут, когда предложение президента станет реальностью и «заработает» новый механизм финансирования. Но времени все меньше: лекарство Матвею нужно вести до двух лет.
А значит, благотворители – пока единственный шанс на спасение. В их поисках старший брат мальчика 11-летний Артём надевает костюм медведя и вместе с папой выходит на улицы Петербурга.

«Надеюсь, что какой-то добрый человек отправит какую-то сумму денег и так по чуть-чуть. Я хочу, чтобы мой брат жил и был здоров», – говорит брат Матвея Артем Куликовский.
За несколько месяцев удалось собрать всего 100 тысяч долларов, но папа малыша продолжает верить… не в чудо – в людей:
«Мир не без добрых людей. И нам помогают. Мы благодарны. Надеемся, что найдется тот волшебник, который подарит жизнь Матвею».

ТОЧНАЯ И ОПЕРАТИВНАЯ ДИАГНОСТИКА – ПУТЬ К СПАСЕНИЮ ЖИЗНИ

Заведующая педиатрическим подразделением трансплантации костного мозга
доктор Ирина Зайдман с маленькой пациенткой
В клинике «Хадасса» используются инновационные методы диагностики орфанных заболеваний:
- усовершенствованные средства компьютерной визуализации
- продвинутая аппаратура
- новейшие тесты.
Во многих случаях орфанные болезни могут быть идентифицированы лишь на основании генетических исследований. На базе клиники «Хадасса» работают ведущие генетики мира. В их распоряжении имеются самые современные тесты, которые обеспечивают не только максимальную точность, но и оперативность диагностики.
Уникальное оборудование

Секвенатор NovaSeq 6000 компании Illumina
В отличие от остальных израильских медицинских центров, где данный прибор используется почти исключительно в исследовательских целях, в клинике «Хадасса» его активно применяют в диагностике генетических заболеваний.
Это различие в подходах обусловлено размером базы генетических данных: при наличии небольшой базы работа NovaSeq 6000, заключающаяся в проведении быстрого сравнительного анализа, теряет смысл. В клинике «Хадасса» собрана самая полная база генетических данных в Израиле, которая называется HADASSOME, поэтому только здесь секвенатор используется в клинических целях. Генетики из других медицинских центров страны постоянно обращаются к иерусалимским коллегам за консультацией.
NovaSeq 6000 значительно снизил стоимость секвенирования экзомов в Израиле
Прибор сканирует ДНК, которую извлекают из крови пациента, полученной в ходе обычного анализа крови. Генетический материал помещают в специальную проточную кювету, разделенную на множество ячеек. К сканированию материал готовит современная роботизированная система.
NovaSeq 6000 обладает повышенной точностью — каждую такую ячейку прибор сканирует 32 млн. раз.
Еще одно преимущество новейшего аппарата — пропускная способность: он может сканировать до 30 генетических проб одновременно.
Секвенирование экзомов проводится с помощью системы самых современных и быстродействующих компьютерных программ под названием PipeLine.
Все эти совершенные технологии позволили не только сделать процесс секвенирования гораздо более точным, но и значительно его ускорить: пациент получает результат меньше чем за 2 недели.
Хадассе нет равных в диагностике орфанных заболеваний
Для обнаружения орфанных заболеваний в медицинском центре используются самые современные генетические тесты, опираясь на которые специалисты иерусалимской больницы безошибочно обнаруживают любые известные генные мутации, при своевременном обращении – на самой ранней стадии. Но это еще не все: врачам Хадассы также нередко удается пополнить длинный список редких болезней.
Так в апреле 2017 года в одном из самых престижных международных изданий в области генетики American Journal of Human Genetics появилась публикация о результатах исследований, проведенных группой ученых под руководством заведующей отделением генетических и метаболических заболеваний МЦ Хадасса профессора Орли Эльпелег. Исследовательская группа сумела впервые описать неизвестное до сих пор опасное генетическое заболевание, поражающее нервную систему ребенка, и разработать методы его раннего обнаружения.
Профессор Орли Эльпелег
Эксперт мирового уровня в области клинической генетики, педиатр, заведующая отделением генетических и метаболических заболеваний МЦ Хадасса.
Благодаря безошибочным диагнозам профессора Эльпелег были спасены сотни детей.
Читать резюме врача Записаться на консультацию
Орфанные заболевания: перечень
В качестве первичной диагностики орфанных заболеваний новорожденные дети проходят специальный неонатальный скрининг согласно приказу Минздрава РФ.
Данный вид обследования направлен на выявление пяти заболеваний, которые можно выявить непосредственно после рождения:
- фенилкетонурия;
- муковисцидоз;
- гипотиреоз;
- галактоземия;
- адреногенитальный синдром.
Внимание! Благодаря процедуре за 10 лет с начала проведения тестов врачам удалось спасти жизни более 10 тысяч детей. Скрининг – это бесплатная процедура, которая выполняется за счет страхового полиса
Проведение исследований на выявление других болезней является платным. Однако Минздрав планирует расширить диагностику, включив в анализ 25 заболеваний
Скрининг – это бесплатная процедура, которая выполняется за счет страхового полиса. Проведение исследований на выявление других болезней является платным. Однако Минздрав планирует расширить диагностику, включив в анализ 25 заболеваний.
БЛАГОТВОРИТЕЛЬНЫЕ ОРГАНИЗАЦИИ
Международный отдел клиники постоянно сотрудничает с благотворительными организациями, оказывая пациентам всевозможное содействие в получении денежной помощи:
- направляет их в фонды;
- предоставляет нужную документацию, медицинские программы и рекомендательные письма от специалистов;
- оказывает поддержку в организации сбора средств.
За последние несколько лет нашим пациентам неоднократно оказывали помощь следующие фонды:
- РУСФОНД;
- WORLD VITA;
- БФ «Алёша»;
- БФ «Артёмка»;
- БФ Помогать легко;
- «Клуб Добряков»;
- Немецкий фонд BILD Ein Herz fur Kinder.
и другие благотворительные организации.
Цена здоровья
Чаще всего орфанные заболевания неизлечимы, но жизнь большинства их носителей сегодня можно облегчить. Правда почти всегда эта помощь стоит больших денег.
В стране часть «орфанников» находится «на содержании» федерального бюджета, за других отвечают регионы. Справляются не все. И тогда людям приходится искать помощь у контролирующих органов и общественных организаций.
«Идём в суды, потому что у них нет денег, не могут принять решение, но чаще это история личности, зависит от участия личности в истории», – рассказывает директор Центра помощи пациентам «Геном» Елена Хвостикова.
У семьи Яковенко из Краснодара путь к жизненно необходимым лекарствам тоже был непростым. Несколько лет обивали пороги врачей и чиновников.

«Стоит это дорого – около 60 миллионов в год. Наш 4 тип единственный, кто на региональном обеспечении. И это страшно, потому что если регион небогатый, то ребенок останется без лечения», – поясняет Клавдия Яковенко.
Не прыгать! Не бегать! Не играть в активные игры! С детьми Клавдии приходится быть строгой:
При рождении Иосиф и Мария казались здоровыми, но с годами их тела стали деформироваться.

Первые диагнозы оказались ошибочными. Дети начали терять зрение и слух. Когда Яковенко узнали причину, их мир навсегда изменился. Мукополисахаридоз IV типа – словно приговор.
«Мукополисахара накапливаются и, если их не выводить, наступает как интоксикация – рушится весь организм. Поэтому существует ферментазаместительная терапия, и чем раньше начать, тем последствий может быть меньше», – рассказывает Яковенко.
В их случае время было упущено. «Выбивать» лекарства пришлось через прокуратуру. Суд вынес решение в пользу семьи. Уже год Иосифа и Марию еженедельно госпитализируют «на капельницы». Прогресс ощутимый. Сейчас Яковенко собираются в очередное путешествие.
Редкие генетические заболевания
Среди всех редких заболеваний большинство приходится на генетические или врожденные болезни, которые имеют хроническое течение и проявляются у детей в разные периоды жизни. Большинство редких генетических патологий манифестируют в младенчестве и сопровождаются расстройствами зрения и слуха, выраженными изменениями внешности, нарушениями двигательной и психической функции. При этом вследствие прогрессивного течения генетической болезни более 25 % детей не доживают до 4-5 лет.
Наследственные заболевания у детей характеризуются неблагоприятным прогнозом. Однако своевременное лечение редких генетических болезней в большинстве случаев дают хорошие результаты, врачам удается улучшить качество жизни маленьких пациентов и увеличить показатели выживаемости.
Лечение без СМС
«Всего одно СМС может спасти жизнь» – такими призывами пестрят интернет и уличные рекламные щиты. «Орфанников» становится больше, а их лечение дорожает. В июне 2020 года Владимир Путин выступил с предложением, которое должно улучшить жизнь носителей редких заболеваний.
При этом действующие меры поддержки обещают сохранить.
«Эти деньги будут предназначены тем детям с тяжелыми хроническими заболеваниями, включая орфанные, помощь которым не подпадает ни под одну из существующих программ медицинской помощи», – говорит Сергей Куцев.
Если говорить только об «орфанниках», то на новый фонд смогут рассчитывать 4,5 тысячи детей. Он – свет в конце тоннеля и для маленького петербуржца Матвея Куликовского.
Ваня выздоровел
В Украине Ване Волохатюку в течение 2 лет не могли поставить диагноз. В МЦ Хадасса у него обнаружили редкое и очень опасное заболевание – синдром IPEX. Ребенка спасли, проведя успешную трансплантацию костного мозга (ТКМ).
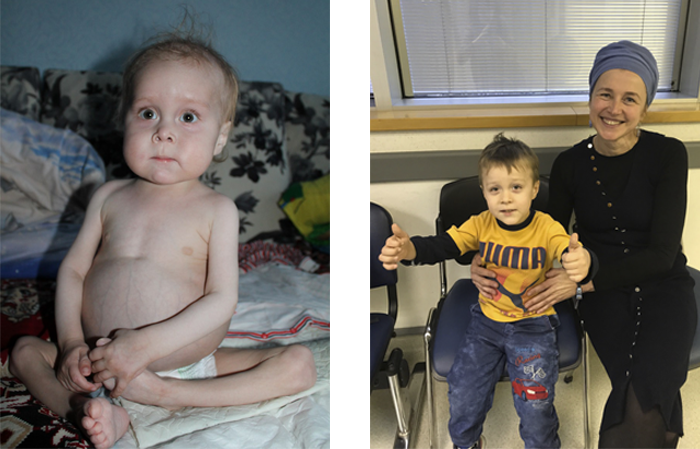
Фото слева: так Ваня выглядел до того, как профессор Полина Степенски успешно провела ему ТКМ в медицинском центре Хадасса.
Фото справа: Ваня после ТКМ с сотрудницей нашего медицинского департамента Эстер Винокуров.
Для справки:
- По разным оценкам в России к 2017 году было зарегистрировано от 1.5 до 5 миллионов больных с редкими заболеваниями.
- В 2017 году в Украине на лечение орфанных заболеваний был выделен 1 млрд. гривен, и эта сумма почти сразу же была признана недостаточной.
- В Беларуси зарегистрированы тысячи случаев орфанных заболеваний, по оценкам местной прессы ими болеют от 6% до 8% населения.
Методы диагностики адреногенитального синдрома
Диагностика в период внутриутробного развития
При обследовании беременных о возможности гиперплазии надпочечников может свидетельствовать аномальное формирование половых органов плода. Это можно увидеть на втором скрининговом акушерском УЗИ. В таких случаях рекомендуется дополнительное обследование, назначается анализ амниотической жидкости или после 21 недели берут кровь плода (из пуповины под контролем УЗИ).
Диагностика после родов
После родов диагностика не представляет сложности. Назначается обследование: проба с АКТГ – нет повышения содержания кортизола после его введения; анализ крови – снижен натрий, повышен калий, 17 ОН-прогестерон в 5-7 раз и более, андростендион (предшественник стероидов), ренин (при сольтеряющей форме); анализ мочи – высокая концентрация 17-кетостероидов, после приема преднизолона снижается наполовину. В сомнительных случаях применяют кариотипирование для подтверждения женского кариотипа (46ХХ). Исследование мутаций в гене CYP11B1 – секвенирование на выявление мутации – His R448H.
Диагностика при позднем клиническом дебюте
Большее значение диагностический поиск приобретает при позднем клиническом дебюте или скрытом течении с минимальными внешними проявлениями вирилизации. В подобных ситуациях для выявления адреногенитального синдрома используют следующие лабораторные и инструментальные методы:
-
гормональный профиль (кортизол и предшественники, 17-ОНП, ДЭА-с и ДЭА, АКТГ и др.);
-
рентгенография костей кисти (раннее завершение роста);
-
УЗИ органов малого таза (признаки ановуляторного цикла), надпочечников (гиперплазии); в ряде случаев назначается МРТ, МСКТ исследование;
-
измерение базальной температуры. Температурная кривая типична для ановуляторного цикла: первая фаза растянута, вторая укорочена, что обусловлено недостаточностью желтого тела, которое не образуется из-за отсутствия овуляции).
Дифференциальная диагностика адреногенитальных расстройств, возникших в пубертатном и детородном возрасте, проводится с синдромом поликистозных яичников, овариальными андробластомами, андростеромами надпочечников, вирильным синдромом гипоталамического происхождения и конституциональным гирсутизмом. В сложных случаях к диагностике привлекают эндокринологов, урологов, врачей-генетиков.
Чтобы точно продиагностировать заболевание, запишитесь на прием к специалистам сети «Семейный доктор».
Диагностика орфанных заболеваний
Генетический анализ на выявление орфанных заболеваний состоит из таких стадий:
- Забор биоматериала.
- Отделение ДНК от сторонних элементов, дальнейшая подготовка образца.
- Оценка ДНК с помощью секвенаторов.
- Анализ полученных результатов посредством диагноза.
Данный процесс может занимать длительный срок (до нескольких лет) и требовать проведения нескольких анализов, чтобы подтвердить диагноз. Кроме того, диагностику могут проводить только специалисты по орфанным заболеваниям, так как многие врачи не сталкиваются с ними в повседневной практике, могут не иметь опыта в данной области.
Один на десять тысяч
Мир полон изученных и неизученных болезней. Примерно 7 тысяч из них – орфанные, то есть редкие. В России это те, что встречаются у одного из десяти тысяч человек.
«У нас есть такие истории, когда люди, не получающие по каким-то причинам лечение, от нас уходят, уходят тяжело, мучительно. Никто не хочет для своих детей этого…» – говоря это, петербурженкаАнна Прохорова с трудом сдерживает волнение. Её дочь в печальной статистике.

Все свои 12 лет Лиза живёт по строгому расписанию: четыре раза в день – ингаляция, питание – по диете. Девочке нужно потреблять 2,5 тысячи килокалорий в сутки – это норма взрослого мужчины. Протеиновый коктейль – как лекарство.
«Калории быстро перевариваются, поэтому нужно больше кушать», – поясняет Елизавета Баскакова.

У Лизы – муковисцидоз. В России этот диагноз – у 3,5 тысяч человек. Генетическое заболевание уничтожает органы пищеварительной системы и легкие.
«Они могут просто отказать: начну задыхаться и меня подключат к аппарату. Если я не буду принимать лекарства и соблюдать терапию, то превращусь в сине-зеленого скелета…» – рассказывает девочка.

Глядя на девочку, в это трудно поверить: модельная внешность и бойкий нрав. Большую часть времени она, как обычный ребенок: ходит в школу, общается с друзьями. А на вопрос: «Чем отличаешься от сверстников?» отшучивается: «Ну, меня не возьмут в космонавты!».
Все меняется, когда происходит обострение. Несколько раз в год без антибиотиков не обойтись. Курс стоит около 300 тысяч рублей. К счастью, финансирует процедуры государство из федерального бюджета, но есть сложности.
Немного о деньгах
Большое количество разработок и постепенная регистрация все большего количества геннотерапевтических препаратов позволяют рассчитывать на то, что на наших глазах рождается фармацевтическая индустрия нового типа.
Главная причина медленного развития направления — сложности в коммерциализации разработок фармацевтическими компаниями. Они не видят рынка, не понимают, как будут окупаться вложения в производство. Но, например, в случае препаратов от GNAO1 эпилепсии на основе олигонуклеотидов стоимость производства одной и 60 доз практически не отличается, а значит, стоимость лечения 1 и 20 пациентов одинакова. Просто за счет диагностики и большего охвата пациентов можно значительно снизить затраты в расчете на одного пациента.
Более того, оборудование и подход являются универсальными, это значит, что не такой уж фантастикой выглядит картина, когда после диагноза в поликлинике пациенты будут становиться в очередь на синтез для них персонального генного препарата. Ведь основным отличием генной терапии от других фармацевтических подходов является то, что мишень известна, известен способ воздействия на нее, а значит, реальный объем затрат на разработку снижается в сотни раз.
Персональные клинические испытания
Во всех случаях возникает совершенно новая проблема обеспечения безопасности лекарственного препарата — обычно на доклинических стадиях исследуется токсичность субстанции, затем безопасность подтверждается на 1-й стадии клинических испытаний на здоровых добровольцах и лишь на 2-й стадии клинических испытаний препарат получают пациенты для исследования его эффективности.
В случае генетических заболеваний на животных зачастую невозможно исследовать прямую генетическую токсичность (нет у животных человеческих генов), а для здоровых добровольцев препарат может быть заведомо опасен. В то же время для орфанных заболеваний невозможно собрать группу для полноценных исследований на больных, если их на всю планету десять человек.
В Институте биологии гена Российской академии наук совместно с компанией «Марлин Биотех» ведется разработка нескольких геннотерапевтических лекарственных средств. Мы ищем лекарство от дистрофии Дюшенна, GNAO1 эпилепсии и FLNC кардиомиопатии. Во всех случаях речь идет о персонализированной терапии.
Общая схема работы по проектам состоит из нескольких этапов — генетическая диагностика, разработка молекулярного подхода к терапии, создание персонализированной животной модели заболевания с теми же мутациями, что обнаружены у пациента, создание лекарственного препарата на основе олигонуклеотидов или минигена, разработка системы их доставки и испытания ее эффективности на культуре клеток, а затем на модельных животных.
Все это позволяет провести комплекс исследований, для того чтобы показать общую, специфическую токсичность потенциального лекарственного препарата, а главное — его эффективность уже на животной модели, для того чтобы у регулятора были основания для разрешения клинических исследований на одном пациенте. Пока ни в России, ни в мире такой механизм не отработан, и в каждом случае требуется индивидуальный подход к исследованиям.
Один из многих тысяч
Значительная часть орфанных заболеваний является не просто генетическими, но моногенными. Для них известна генетическая мишень, на коррекцию дефекта которого должна быть направлена генная терапия. Вот только один пример.
Мутации в гене GNAO1 приводят к эпилептической энцефалопатии с двигательными нарушениями, грубой задержке психомоторного развития. На практике это приводит к тому, что у детей присутствует, в зависимости от варианта мутации, эпилепсия, хорея, гиперкинезы, а также дети не держат голову, не сидят, не разговаривают. При этом, по мнению неврологов, у детей сохраняется интеллект.
Есть два вида мутаций, при которых присутствуют все симптомы: R209H и G203R, таких детей в мире сейчас более 200. Второй вариант наиболее тяжелый и приводит ко всем перечисленным выше симптомам, с ним нет ни одного ребенка старше 8 лет. На сегодняшний день в мире диагностировано 24 ребенка с наиболее тяжелым вариантом мутации, пять из них в России. Генная терапия, направленная на подавление работы мутантного гена, — единственный шанс выжить для этих пациентов.
Причины адреногенитального синдрома
В 90-95% случаев патология возникает при повреждении активного гена CYP21-B, который отвечает за синтез 21-гидроксилазы – фермента, влияющего на образование кортизола. В остальных клинических случаях АГС развивается вследствие дефектов ДНК, нарушающих производство других ферментов, обеспечивающих стероидогенез, – StAR/20,22-десмолазы, 3-β- гидрокси-стероиддегидрогеназы, 17-α-гидроксилазы/17,20-лиазы, 11-β-гидроксилазы, P450-оксидоредуктазы и синтетазы альдостерона.
Различают следующие виды адреногенитального синдрома:
-
сольтеряющий тип. Проявления болезни начинают обнаруживаться с первых недель жизни. У девочек половые органы формируются по мужскому типу, а у мальчиков увеличиваются пенис и мошонка. Из-за грубого нарушения образования стероидных гормонов развивается тяжелая рвота, понос, судороги, темнеет кожа. Прогрессирующее обезвоживание ведет к смерти при отсутствии своевременной заместительной терапии.
Через 2-3 дня от начала клинического проявления заболевания появляются симптомы дегидратации и развития метаболического ацидоза.
-
Простой вирильный. Преобладают нарушения развития половых органов. У девочек: увеличен клитор, похожий на половой член; углублен вход во влагалище; половые губы больше нормы; матка и придатки сформированы. У мальчиков: увеличен пенис, кожа мошонки имеет темную пигментацию. Девочки широкоплечие, с узким тазом, укороченными и массивными конечностями. У них низкий голос, кадык на шее, молочные железы не растут. У мальчиков рано появляются грубые волосы на подбородке и верхней губе, ломается голос.
С возрастом у пациентов нарастают признаки вирилизации вследствие стимулирующего действия андрогенов. Характерно ранее начало полового созревания в 3-5 лет.
-
Постпубертатный тип. Выявляют в молодом возрасте. Приобретенная форма возникает после появления опухоли в надпочечниках или при их повышенной активности.
Половые органы соответствуют полу, но развивается гипертрофия клитора и пениса. Основные нарушения встречаются у женщин – уменьшаются или прекращаются месячные (после стресса, травмы, аборта, выкидыша). Наступление первой менструации может быть только к 15 годам, цикл удлинен (более 30-40 дней). Кожа склонна к повышенной жирности и образованию угрей, имеется повышенное оволосение. При обследовании обнаруживают гиперплазию коркового слоя надпочечников.
Также имеются и варианты неклассического течения адреногенитального синдрома – с высоким давлением крови, с лихорадкой, липидный (ожирение, нарушение холестеринового обмена), с гирсутизмом (усиленный рост волос на теле и лице, по средней линии живота, около сосков).
Лечение редких заболеваний (методы)
Большая часть редких заболеваний — это неизлечимые тяжелые патологии, угрожающие жизни. Но сегодня существует ряд эффективных методов, позволяющих поддерживать нормальное качество жизни, а также достигать выживаемости в течение многолетнего периода. На сегодняшний день разработано около 450 лекарственных средств, устраняющих одно из звеньев патогенеза. Орфанные препараты применяют для лечения редких заболеваний, которые могут привести к инвалидности или угрожают жизни пациента.
Более 95 % редких заболеваний не лечатся. Однако генетики сегодня проводят доклинические исследования по новым технологиям лечения онкологических и генетических патологий. Это методы РНК-интерференции и редактирование генома.
Диагностика и лечение редких заболеваний в Москве
Из-за отсутствия лабораторно-технической базы и достаточного числа квалифицированных специалистов во многих регионах, российские пациенты нередко сталкиваются с проблемой своевременной и точной диагностики редких заболеваний. Однако сегодня в стране функционируют несколько федеральных медцентров, в том числе и в Москве, где есть все условия для проведения тщательной диагностики при подозрениях на орфанные болезни.
Сейчас каждый ребенок после рождения проходит неонатальный скрининг на более 5 редких заболеваний, включая генетические патологии. Верификация диагноза выполняется ведущими генетиками в современных лабораториях.
Вопрос лечения редких болезней является нерешенным на уровне отечественной медицины, поскольку большинству орфанных пациентов необходимые оригинальные препараты не доступны. В России занесено в Федеральный реестр около 17. тыс. человек, которые находятся под постоянным наблюдением специалистов. Если для редкой болезни не существует эффективного лечения, врачи поддерживают состояние больных и увеличивают продолжительность их жизни при помощи специализированного питания и орфанных препаратов.
Диагностика и лечение редких заболеваний в Израиле
Для выявления редких заболеваний специалисты Израиля используют инновационные генетические тесты, безошибочно обнаруживая известные мутации на ранней стадии развития. Ведущие эксперты в данной области медицины не только устанавливают точный диагноз, но и лечат орфанные болезни, применяя новейшие методы энзимозаместительной и генной терапии. Исключительную эффективность при многих генетических патологиях доказала трансплантация костного мозга, проводимая всемирно известными гематологами и хирургами Израиля.
Орфанные заболевания: статистика
Для каждой страны оценка «редкости» заболевания сугубо индивидуальна.
Например, в Европе установлено отношение одного случая орфанного заболевания к 2 тыс. При этом количество пациентов там составляет более 30 млн. человек.
В России действует другое соотношение: 10 на 100 тыс. граждан.
Согласно данным Росстата, 15,8 тыс. россиян болеют редкими заболеваниями, более 7 тыс. из них – дети. В то же время Министерство здравоохранения России учитывает не все случаи заболеваний, а только те, которые подтверждаются покупкой лекарственных средств в рамках нескольких программ:
- В региональную программу входит факт выявления орфанных заболеваний, угрожающих жизни ребенка, оказание финансовой помощи из бюджета.
- В федеральную программу входит выявление и оказание помощи тем пациентам, лечение которых является дорогостоящим. К таким случаям можно отнести заболевания рассеянным склерозом, нанизмом, болезнью Гоше и т.д.
Важно! В отдельную категорию следует отнести лиц, нуждающихся в трансплантации внутренних органов
Редкие орфанные заболевания
В рамках государственного задания от Федерального агентства научных организаций с 2007 года в России установлена отдельная квота на обследование и консультацию 10 тыс. пациентов, страдающих орфанными болезнями.
Несмотря на то, что Министерство здравоохранения уделяет особое внимание людям с орфанными заболеваниями (в том числе, выявляет их наличие и оказывает помощь в предоставлении специальных оборудования и препаратов для лечения)
Вопрос денег
У сына Натальи Буртаевой мукополисахаридоз IV типа, заболевание схожее, но ни в списке «12 нозологий», ни в «Перечне 24» его нет.
Даниилу уже шестнадцать. Его рост 97 сантиметров. Ходит с трудом: ноги и руки деформированы, внутренние органы тоже деформированы. С пятого класса Даня учится на дому. Нервная система при этом диагнозе не страдает, дети с ним отлично осваивают школьную программу. Но поврежденной оказывается опорно-двигательная система, страдает слух.
Препараты специальной ферментной заместительной терапии, положенной в таких случаях, существуют, но первое время были не зарегистрированы в России. Единственное лекарство, одобренное американским надзорным фармакологическим ведомством (FDA) в 2014 году, называется элосульфаза альфа — торговое название Вимизим (Vimizim).
Без Вимизима больные мукополисахаридозом IV типа умирают к двадцати годам. Когда новости о его разработке только появились в специализированных СМИ, Наталья было обрадовалась. Но стоимость его годового курса превышает 60 миллионов рублей.
Минздрав Ульяновской области ожидаемо отказался приобретать препарат. Наталья подала в суд и выиграла: первый в 2016 году, второй, когда министерство подало апелляцию, — в 2017. Ни письма в Росздравнадзор и Путину, ни сюжет на «Первом канале» не помогли: Даниил до сих пор не получает лечения.
«Мы не прячемся, — твердо сообщает Наталья корреспонденту. — Даня часто гуляет на коляске. Мы с ним ездили на море, а сегодня вообще особенный день: последний звонок. Он выйдет на сцену и получит аттестат зрелости.
Добавляет: «Он у меня обычно говорит, когда я переживаю, что люди его не примут: „Мам, ты, это, не переживай. Я привык, что на меня так смотрят“».
Более того, после школы Даниил планирует учиться дальше, признается мать, вздыхая: «Не знаю, сколько он будет жить». Врачи точно ответить на этот вопрос тоже не в силах, но очевидно, что без терапии Даня проживет недолго.
Примеры орфанных заболеваний, которые диагностируют и лечат в МЦ Хадасса
- геморрагическая тромбоцитемия
- синдром Имерслунда-Грасбека
- альфа-талассемия
- бета-талассемия
- дельта-бета-талассемия
- серповидно-клеточная анемия
- наследственный сфероцитоз
- наследственный эллиптоцитоз
- гемолитическая анемия
- атипичный гемолитико-уремический синдром
- апластическая анемия
- анемия Даймонда-Блекфена
- пурпура
- гипергаммаглобулинемия
- IPEX-синдром
- аденозиндезаминазная недостаточность
- недостаточность лейкоцитарной адгезии
- ретикулярный дисгенез
- болезнь Виллебранда
- детский генетический агранулоцитоз
- врожденная метгемоглобинемия
- эозинофилия
- гемофагоцитарный лимфогистиоцитоз
- синдром Розаи-Дорфмана
- комбинированный иммунодефицит
- синдром Вискотта-Олдрича
- синдром Ниймаген
- синдром Блума
- хроническая гранулематозная болезнь (ХГБ)
- синдром ICF: NEMO синдром
- синдром Ди Георга (Ди Джорджа, велокардиофациальный синдром)
- синдром Оменна
- синдром Чедиака-Хигаси
- хрящево-волосяная гипоплазия
Метаболические заболевания и болезни эндокринной системы
- детская пролактинома
- болезнь Иценко-Кушинга
- адреногенитальный синдром
- синдром Ларона
- врожденный гиперинсулинизм
- тирозинемия
- болезнь «кленового сиропа»
- адренолейкодистрофия
- синдром Фанкони
- цистиноз
- болезнь Хартнупа
- синдром Лоу
- гликогеноз
- галактоземия
- болезнь Гоше
- болезнь Фабри
- болезнь Ниманна-Пика
- муковисцидоз
- семейная средиземноморская лихорадка
- аспартилглюкозаминурия
- синдром Вольфрама
- болезнь Фарбера
- метахроматическая лейкодистрофия
- мукополисахаридоз 1 типа (синдром Гурлера-Шейе)
- мукополисахаридозы III, IV, VI, VII типов
- муколипидоз
- альфа-маннозидоз
- бета-маннозидоз
- сиалидоз
- галактосиалидоз
- а-бета-липопротеинемия
- синдром Леша-Нихана
- болезнь Менкеса («болезнь курчавых волос»)
- порфирия
- первичный ювенильный гемохроматоз
- болезнь Александера
- болезнь Краббе
- синдром Прадера–Вилли
- синдром Швахмана-Даймонда
Заболевания нервной системы
- синдром Туретта
- болезнь Галлервордена-Шпатца
- дистрофия Дюшенна
- мальформация Арнольда-Киари тип 1
- сирингомиелия
Врожденные аномалии
- конгенитальный дискератоз
- остеопетроз (мраморная болезнь)
- синдром Энгельмана
- болезнь Гиршпрунга
- синдром Альпорта
- нейрофиброматоз 1 типа
- синдром Мебиуса
- синдром Гольденхара
- синдром Холта-Орама
- синдром Лорена–Муна-Бидля
- туберозный склероз (болезнь Бурневилля)
- синдром Пьера-Робена
- дисплазическая дистрофия
- врожденный ихтиоз